Load packages and data
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
library(tidyr)
library(limma)
library(splines)
library(MSMICA)
# this is the working directory for the case study
setwd('/Users/james/Desktop/Emory University - Ph.D./PhD dissertation/MSMICA/Publication/Abstract/MSMICA_package/MSMICA/vignettes')
# import feature table data
## hilic positive data
data(feature_table_exp_hilicpos)
print(feature_table_exp_hilicpos)
## # A tibble: 11,267 × 44
## mz time NM5_GS_0_1 NM5_GS_0_3 NM5_GS_0_5 TM5_GS_0_1 TM5_GS_0_3 TM5_GS_0_5
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 85.0 49.4 127294848 93929747 66033558 159310776 99288246 82335881
## 2 85.0 83.4 9282517 14636989 12921267 8682974 10271317 7132031
## 3 85.1 109. 13329008 15703815 8642594 8449978 8644413 3355450
## 4 85.2 30.8 0 26889920 32196356 25253361 47516277 54826651
## 5 85.3 31.5 0 8566404 14599669 9787948 14132846 23874217
## 6 85.4 30.2 0 13508765 16721105 61654938 9957638 3201424
## 7 85.4 33.8 15066696 0 0 11540798 20251857 31230557
## 8 85.6 156. 6775208 4078654 0 2803849 107620 0
## 9 85.6 76.0 0 0 0 4678248 3290240 1366488
## 10 85.8 106. 0 69397733 0 0 0 0
## # ℹ 11,257 more rows
## # ℹ 36 more variables: NM11_GS_0_1 <dbl>, NM11_GS_0_3 <dbl>, NM11_GS_0_5 <dbl>,
## # TM11_GS_0_1 <dbl>, TM11_GS_0_3 <dbl>, TM11_GS_0_5 <dbl>, NM6_GS_0_1 <dbl>,
## # NM6_GS_0_3 <dbl>, NM6_GS_0_5 <dbl>, TM4_GS_0_1 <dbl>, TM4_GS_0_3 <dbl>,
## # TM4_GS_0_5 <dbl>, NM4_GS_0_1 <dbl>, NM4_GS_0_3 <dbl>, NM4_GS_0_5 <dbl>,
## # TM6_GS_0_1 <dbl>, TM6_GS_0_3 <dbl>, TM6_GS_0_5 <dbl>, NM7_GS_0_1 <dbl>,
## # NM7_GS_0_3 <dbl>, NM7_GS_0_5 <dbl>, NM9_GS_0_1 <dbl>, NM9_GS_0_3 <dbl>, …
## c18 negative data
data(feature_table_exp_c18neg)
print(feature_table_exp_c18neg)
## # A tibble: 8,456 × 44
## mz time NM5_GS_0_2 NM5_GS_0_4 NM5_GS_0_6 TM5_GS_0_2 TM5_GS_0_4 TM5_GS_0_6
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 85.0 123. 3451142 17675474 4332024 8899759 12204896 12666665
## 2 85.0 288. 2267915 3117932 3828736 4224009 4200063 3788690
## 3 85.0 293. 3691321 2391226 3155868 2361909 4033037 2426635
## 4 85.0 24.9 38858050 43782168 32818312 36913236 36235779 28511185
## 5 85.0 95.0 3104800 2580525 3767773 2646283 2663336 1084595
## 6 85.4 236. 6096726 4051104 1465424 5114500 971445 2515400
## 7 85.5 22.0 0 0 0 0 0 0
## 8 85.5 210. 0 2349999 5895828 1703156 3593730 8463200
## 9 85.6 211. 0 258962 2349208 0 1626943 3316184
## 10 85.7 217. 747185 3036484 5062762 4169043 3809372 9121575
## # ℹ 8,446 more rows
## # ℹ 36 more variables: NM11_GS_0_2 <dbl>, NM11_GS_0_4 <dbl>, NM11_GS_0_6 <dbl>,
## # TM11_GS_0_2 <dbl>, TM11_GS_0_4 <dbl>, TM11_GS_0_6 <dbl>, NM6_GS_0_2 <dbl>,
## # NM6_GS_0_4 <dbl>, NM6_GS_0_6 <dbl>, TM4_GS_0_2 <dbl>, TM4_GS_0_4 <dbl>,
## # TM4_GS_0_6 <dbl>, NM4_GS_0_2 <dbl>, NM4_GS_0_4 <dbl>, NM4_GS_0_6 <dbl>,
## # TM6_GS_0_2 <dbl>, TM6_GS_0_4 <dbl>, TM6_GS_0_6 <dbl>, NM7_GS_0_2 <dbl>,
## # NM7_GS_0_4 <dbl>, NM7_GS_0_6 <dbl>, NM9_GS_0_2 <dbl>, NM9_GS_0_4 <dbl>, …
# replace all 0 values with NA after column 2
feature_table_exp_hilicpos[, -c(1:2)] = feature_table_exp_hilicpos[, -c(1:2)] %>%
mutate_all(~na_if(., 0))
# replace all 0 values with NA after column 2
feature_table_exp_c18neg[, -c(1:2)] = feature_table_exp_c18neg[, -c(1:2)] %>%
mutate_all(~na_if(., 0))
# median summarize the triplicates
df <- feature_table_exp_hilicpos
# Gather the triplicates into a long format
df_long <- df %>%
pivot_longer(cols = -c(mz, time), names_to = "sample", values_to = "value")
# Extract the base sample name (without the replicate number)
df_long <- df_long %>%
mutate(sample_base = sub("(_[0-9])$", "", sample))
# Compute the median for each set of triplicates
df_median <- df_long %>%
group_by(mz, time, sample_base) %>%
summarize(median_value = median(value), .groups = "drop")
# Pivot back to the wide format
feature_table_exp_hilicpos <- df_median %>%
pivot_wider(names_from = sample_base, values_from = median_value)
# View the summarized data frame
print(feature_table_exp_hilicpos)
## # A tibble: 11,267 × 16
## mz time NM11_GS_0 NM4_GS_0 NM5_GS_0 NM6_GS_0 NM7_GS_0 NM8_GS_0 NM9_GS_0
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 85.0 49.4 91637758 53374857 93929747 47592345 49580672 65890518 24709252
## 2 85.0 83.4 20848423 11877054 12921267 17645801 7985920 17303431 10586069
## 3 85.1 109. 12062332 3298088 13329008 9183408 1647494 15424423 10908045
## 4 85.2 30.8 43116622 24855153 NA 8517991 34448219 29816051 24415023
## 5 85.3 31.5 13212572 10524120 NA NA 16114654 NA NA
## 6 85.4 30.2 NA NA NA 5593932 9671769 NA 11869357
## 7 85.4 33.8 NA 11456826 NA NA 8251705 NA NA
## 8 85.6 156. NA NA NA 2907723 NA NA NA
## 9 85.6 76.0 NA NA NA NA NA 3804028 NA
## 10 85.8 106. NA NA NA NA NA NA NA
## # ℹ 11,257 more rows
## # ℹ 7 more variables: TM11_GS_0 <dbl>, TM4_GS_0 <dbl>, TM5_GS_0 <dbl>,
## # TM6_GS_0 <dbl>, TM7_GS_0 <dbl>, TM8_GS_0 <dbl>, TM9_GS_0 <dbl>
# median summarize the triplicates
df <- feature_table_exp_c18neg
# Gather the triplicates into a long format
df_long <- df %>%
pivot_longer(cols = -c(mz, time), names_to = "sample", values_to = "value")
# Extract the base sample name (without the replicate number)
df_long <- df_long %>%
mutate(sample_base = sub("(_[0-9])$", "", sample))
# Compute the median for each set of triplicates
df_median <- df_long %>%
group_by(mz, time, sample_base) %>%
summarize(median_value = median(value), .groups = "drop")
# Pivot back to the wide format
feature_table_exp_c18neg <- df_median %>%
pivot_wider(names_from = sample_base, values_from = median_value)
# View the summarized data frame
print(feature_table_exp_c18neg)
## # A tibble: 8,456 × 16
## mz time NM11_GS_0 NM4_GS_0 NM5_GS_0 NM6_GS_0 NM7_GS_0 NM8_GS_0 NM9_GS_0
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 85.0 123. 7722094 10229284 4332024 5469528 8399260 2175857 1929214
## 2 85.0 288. 4407084 3951785 3117932 3267173 4019103 3336862 3128299
## 3 85.0 293. 2607157 3002005 3155868 1907645 1002345 825507 2679529
## 4 85.0 24.9 64373076 39894574 38858050 35446680 35724755 34192423 33235053
## 5 85.0 95.0 2999487 2543380 3104800 1624592 2369531 4524972 2823455
## 6 85.4 236. 4240866 4900397 4051104 3749169 2528974 2431985 4520111
## 7 85.5 22.0 NA NA NA NA NA NA NA
## 8 85.5 210. 3676008 NA NA 1146822 4697878 6359702 3184766
## 9 85.6 211. 2509843 NA NA NA NA 2801030 NA
## 10 85.7 217. 4761129 3545481 3036484 1748788 3907803 7352392 3096899
## # ℹ 8,446 more rows
## # ℹ 7 more variables: TM11_GS_0 <dbl>, TM4_GS_0 <dbl>, TM5_GS_0 <dbl>,
## # TM6_GS_0 <dbl>, TM7_GS_0 <dbl>, TM8_GS_0 <dbl>, TM9_GS_0 <dbl>
# Filter out features appear less than 20% of all samples. The intensity column starts from the 3rd column.
feature_table_exp_hilicpos = QC_filter(x = feature_table_exp_hilicpos, metabolite_start_column = 3, minimum_sample_appear = 0.20)
feature_table_exp_c18neg = QC_filter(x = feature_table_exp_c18neg, metabolite_start_column = 3, minimum_sample_appear = 0.20)
# import MSMICA results
## hilic positive data
hilicpos_MSMICA_identified_metabolites = read_csv("MSMICA_test_hilicpos_MSMICA_identified_metabolites_filtered.csv")
## Rows: 2926 Columns: 73
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: ","
## chr (19): mz_time, Adduct, Name, identification_type, identification_method,...
## dbl (53): mz, time, Probability, metabolite_within_feature_number, time_pred...
## lgl (1): isomer_exist
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
print(hilicpos_MSMICA_identified_metabolites)
## # A tibble: 2,926 × 73
## mz time mz_time Adduct Name identification_type identification_method
## <dbl> <dbl> <chr> <chr> <chr> <chr> <chr>
## 1 348. 284 348.0703_… M+H AMP Schymanski level 3a mz matching; retenti…
## 2 385. 90 385.1282_… M+H S-Ad… Schymanski level 3a mz matching; retenti…
## 3 148. 78 148.0604_… M+H Glut… Schymanski level 3a mz matching; retenti…
## 4 181. 37 181.072_37 M+H D-Gl… Schymanski level 3a mz matching; retenti…
## 5 90.1 70 90.055_70 M+H Alan… Schymanski level 3a mz matching; retenti…
## 6 608. 120 608.0855_… M+H Urid… Schymanski level 3b mz matching; retenti…
## 7 147. 110 147.1128_… M+H Lysi… Schymanski level 3a mz matching; retenti…
## 8 134. 83 134.0448_… M+H Aspa… Schymanski level 3a mz matching; retenti…
## 9 308. 225 308.0909_… M+H Glut… Schymanski level 3a mz matching; retenti…
## 10 324. 34 324.059_34 M+H CMP Schymanski level 3a mz matching; retenti…
## # ℹ 2,916 more rows
## # ℹ 66 more variables: Probability <dbl>,
## # metabolite_within_feature_number <dbl>, isomer_exist <lgl>,
## # time_predicted <dbl>, time_difference <dbl>, mean_intensity <dbl>,
## # PubChem_compound_id <dbl>, HMDB_ID <chr>, KEGG_ID <chr>,
## # LIPID_MAPS_ID <chr>, ChEBI_ID <dbl>, BioCyc_ID <chr>, DrugBank_ID <chr>,
## # Mono_mass <dbl>, Formula <chr>, Charge <dbl>, SMILES <chr>, …
## c18 negative data
c18neg_MSMICA_identified_metabolites = read_csv("MSMICA_test_c18neg_MSMICA_identified_metabolites_filtered.csv")
## Rows: 2439 Columns: 73
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: ","
## chr (19): mz_time, Adduct, Name, identification_type, identification_method,...
## dbl (53): mz, time, Probability, metabolite_within_feature_number, time_pred...
## lgl (1): isomer_exist
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
print(c18neg_MSMICA_identified_metabolites)
## # A tibble: 2,439 × 73
## mz time mz_time Adduct Name identification_type identification_method
## <dbl> <dbl> <chr> <chr> <chr> <chr> <chr>
## 1 662. 20 662.1014_… M-H NAD Schymanski level 3a mz matching; retenti…
## 2 664. 20 664.1174_… M-H NADH Schymanski level 3a mz matching; retenti…
## 3 742. 20 742.0704_… M-H NADP Schymanski level 3a mz matching; retenti…
## 4 426. 22 426.0222_… M-H ADP Schymanski level 3a mz matching; retenti…
## 5 766. 20 766.1058_… M-H Coen… Schymanski level 3a mz matching; retenti…
## 6 784. 20 784.1525_… M-H FAD Schymanski level 3a mz matching; retenti…
## 7 346. 21 346.0557_… M-H AMP Schymanski level 3a mz matching; retenti…
## 8 383. 27 383.1142_… M-H S-Ad… Schymanski level 3a mz matching; retenti…
## 9 87.0 24 87.0087_24 M-H Pyru… Schymanski level 3a mz matching; retenti…
## 10 808. 21 808.1197_… M-H Acet… Schymanski level 3a mz matching; retenti…
## # ℹ 2,429 more rows
## # ℹ 66 more variables: Probability <dbl>,
## # metabolite_within_feature_number <dbl>, isomer_exist <lgl>,
## # time_predicted <dbl>, time_difference <dbl>, mean_intensity <dbl>,
## # PubChem_compound_id <dbl>, HMDB_ID <chr>, KEGG_ID <chr>,
## # LIPID_MAPS_ID <chr>, ChEBI_ID <dbl>, BioCyc_ID <chr>, DrugBank_ID <chr>,
## # Mono_mass <dbl>, Formula <chr>, Charge <dbl>, SMILES <chr>, …
# Prepare the data for MSMICA targeted metabolomics analysis ############################################################################################################
# to make sure there is no duplicate metabolites with the same name but multiple peaks, group by Name and select the metabolites with highest mean intensity
hilicpos_MSMICA_identified_metabolites = hilicpos_MSMICA_identified_metabolites %>%
group_by(Name) %>%
slice_max(mean_intensity) %>%
ungroup()
c18neg_MSMICA_identified_metabolites = c18neg_MSMICA_identified_metabolites %>%
group_by(Name) %>%
slice_max(mean_intensity) %>%
ungroup()
# transpose the table starting at column 32
hilicpos_MSMICA_featuretable_clean_t = t(hilicpos_MSMICA_identified_metabolites[, 32:ncol(hilicpos_MSMICA_identified_metabolites)])
c18neg_MSMICA_featuretable_clean_t = t(c18neg_MSMICA_identified_metabolites[, 32:ncol(c18neg_MSMICA_identified_metabolites)])
# convert the table to a data frame
hilicpos_MSMICA_featuretable_clean_t = as.data.frame(hilicpos_MSMICA_featuretable_clean_t)
c18neg_MSMICA_featuretable_clean_t = as.data.frame(c18neg_MSMICA_featuretable_clean_t)
# set the column names as the Identified_Name
colnames(hilicpos_MSMICA_featuretable_clean_t) = hilicpos_MSMICA_identified_metabolites$Name
colnames(c18neg_MSMICA_featuretable_clean_t) = c18neg_MSMICA_identified_metabolites$Name
# add Group_ID as a column
hilicpos_MSMICA_featuretable_clean_t$Group_ID = rownames(hilicpos_MSMICA_featuretable_clean_t)
c18neg_MSMICA_featuretable_clean_t$Group_ID = rownames(c18neg_MSMICA_featuretable_clean_t)
# move Group_ID as the first column
hilicpos_MSMICA_featuretable_clean_t = tibble(hilicpos_MSMICA_featuretable_clean_t) %>%
select(Group_ID, everything())
c18neg_MSMICA_featuretable_clean_t = tibble(c18neg_MSMICA_featuretable_clean_t) %>%
select(Group_ID, everything())
# Seperate the Group_ID into columns: Group, Temperature, Time, SampleID
hilicpos_MSMICA_featuretable_clean_t = hilicpos_MSMICA_featuretable_clean_t %>%
separate(Group_ID, c("Group", "Temperature", "Time", "Triplicate"), sep = "_") %>%
# create a new column, sample_tyoe, to indicate the sample type: if Group starts with NM, then it is normal, otherwise it is cancer
mutate(sample_type = ifelse(stringr::str_detect(Group, "^NM"), "normal", "cancer")) %>%
# move sample type to the first column
select(sample_type, everything())
c18neg_MSMICA_featuretable_clean_t = c18neg_MSMICA_featuretable_clean_t %>%
separate(Group_ID, c("Group", "Temperature", "Time", "Triplicate"), sep = "_") %>%
# create a new column, sample_tyoe, to indicate the sample type: if Group starts with NM, then it is normal, otherwise it is cancer
mutate(sample_type = ifelse(stringr::str_detect(Group, "^NM"), "normal", "cancer")) %>%
# move sample type to the first column
select(sample_type, everything())
# separate the Group column into Group and ParticipantID: the first two characters are Group, the rest are ParticipantID
hilicpos_MSMICA_featuretable_clean_t = hilicpos_MSMICA_featuretable_clean_t %>%
separate(Group, c("Group", "ParticipantID"), sep = 2)
c18neg_MSMICA_featuretable_clean_t = c18neg_MSMICA_featuretable_clean_t %>%
separate(Group, c("Group", "ParticipantID"), sep = 2)
# Convert all intensity values to log2
hilicpos_MSMICA_featuretable_clean_t[, -c(1:6)] = hilicpos_MSMICA_featuretable_clean_t[, -c(1:6)] %>%
mutate_all(~log2(.))
c18neg_MSMICA_featuretable_clean_t[, -c(1:6)] = c18neg_MSMICA_featuretable_clean_t[, -c(1:6)] %>%
mutate_all(~log2(.))
# check the data ready for MSMICA targeted metabolomics analysis
print(hilicpos_MSMICA_featuretable_clean_t)
## # A tibble: 42 × 2,929
## sample_type Group ParticipantID Temperature Time Triplicate
## <chr> <chr> <chr> <chr> <chr> <chr>
## 1 normal NM 5 GS 0 1
## 2 normal NM 5 GS 0 3
## 3 normal NM 5 GS 0 5
## 4 cancer TM 5 GS 0 1
## 5 cancer TM 5 GS 0 3
## 6 cancer TM 5 GS 0 5
## 7 normal NM 11 GS 0 1
## 8 normal NM 11 GS 0 3
## 9 normal NM 11 GS 0 5
## 10 cancer TM 11 GS 0 1
## # ℹ 32 more rows
## # ℹ 2,923 more variables:
## # `((2-Amino-3-((2-amino-3-((carboxymethyl)amino)-3-oxopropyl)dithio)propanoyl)amino)acetic acid` <dbl>,
## # `(+)-3-Methoxymorphinan` <dbl>, `(+)-Bornyl-diphosphate` <dbl>,
## # `(+)-Calycanthidine` <dbl>, `(+)-Chebulic acid` <dbl>,
## # `(+)-Dukunolide D` <dbl>, `(+)-Luguine` <dbl>,
## # `(+-)-3-Methyladipic acid` <dbl>, `(+-)-Asarinol B` <dbl>, …
print(c18neg_MSMICA_featuretable_clean_t)
## # A tibble: 42 × 2,445
## sample_type Group ParticipantID Temperature Time Triplicate
## <chr> <chr> <chr> <chr> <chr> <chr>
## 1 normal NM 5 GS 0 2
## 2 normal NM 5 GS 0 4
## 3 normal NM 5 GS 0 6
## 4 cancer TM 5 GS 0 2
## 5 cancer TM 5 GS 0 4
## 6 cancer TM 5 GS 0 6
## 7 normal NM 11 GS 0 2
## 8 normal NM 11 GS 0 4
## 9 normal NM 11 GS 0 6
## 10 cancer TM 11 GS 0 2
## # ℹ 32 more rows
## # ℹ 2,439 more variables:
## # `(+)-(3S,4R)-3,4-dihydroxy-3,4-dihydrofluorene` <dbl>,
## # `(+)-8'-Hydroxyabscisic acid` <dbl>, `(+)-Calycanthidine` <dbl>,
## # `(+)-Eudesmin` <dbl>, `(+)-Plicamine` <dbl>, `(+)-Prosopinine` <dbl>,
## # `(+)-Soyacerebroside I` <dbl>, `(+-)-3-Methyladipic acid` <dbl>,
## # `(-)-2-Aminobutyric acid` <dbl>, `(-)-trans-Carveol glucoside` <dbl>, …
# save the results as a csv file
write.csv(hilicpos_MSMICA_featuretable_clean_t, "MSMICA targeted metabolomics analysis/hilicpos_MSMICA_featuretable_clean_transposed.csv", row.names = FALSE)
write.csv(c18neg_MSMICA_featuretable_clean_t, "MSMICA targeted metabolomics analysis/c18neg_MSMICA_featuretable_clean_transposed.csv", row.names = FALSE)
############################# limma paired t-test #############################
# Factorize the samples into normal and cancer
Group_hilicpos = factor(hilicpos_MSMICA_featuretable_clean_t$sample_type, levels = c("normal", "cancer"))
Group_c18neg = factor(c18neg_MSMICA_featuretable_clean_t$sample_type, levels = c("normal", "cancer"))
# Factorize the ParticipantID
ParticipantID_hilicpos = factor(hilicpos_MSMICA_featuretable_clean_t$ParticipantID)
ParticipantID_c18neg = factor(c18neg_MSMICA_featuretable_clean_t$ParticipantID)
# Create the design matrix
design_T_hilicpos <- model.matrix(~ ParticipantID_hilicpos+Group_hilicpos)
design_T_c18neg <- model.matrix(~ ParticipantID_c18neg+Group_c18neg)
# Extract the intensity values for the limma analysis
hilicpos_intensity = hilicpos_MSMICA_featuretable_clean_t[, 7:ncol(hilicpos_MSMICA_featuretable_clean_t)]
c18neg_intensity = c18neg_MSMICA_featuretable_clean_t[, 7:ncol(c18neg_MSMICA_featuretable_clean_t)]
hilicpos_intensity = as.data.frame(hilicpos_intensity)
c18neg_intensity = as.data.frame(c18neg_intensity)
# transpose the metabolite intensity data
y_T_hilicpos = t(hilicpos_intensity)
y_T_c18neg = t(c18neg_intensity)
# Fit the linear model
fit_hilicpos = lmFit(y_T_hilicpos, design_T_hilicpos)
## Warning: Partial NA coefficients for 255 probe(s)
fit_c18neg = lmFit(y_T_c18neg, design_T_c18neg)
## Warning: Partial NA coefficients for 174 probe(s)
fit2_hilicpos <- eBayes(fit_hilicpos)
fit2_c18neg <- eBayes(fit_c18neg)
# Extract all relevant values from the limma output
limma_outcome_hilicpos = tibble(
Cancer_log2FC = c(fit2_hilicpos$coefficients[, "Group_hilicposcancer"]),
t_statistic = c(fit2_hilicpos$t[, "Group_hilicposcancer"]),
p = c(fit2_hilicpos$p.value[, "Group_hilicposcancer"]))
limma_outcome_c18neg = tibble(
Cancer_log2FC = c(fit2_c18neg$coefficients[, "Group_c18negcancer"]),
t_statistic = c(fit2_c18neg$t[, "Group_c18negcancer"]),
p = c(fit2_c18neg$p.value[, "Group_c18negcancer"]))
limma_outcome_hilicpos$p.fdr = p.adjust(limma_outcome_hilicpos$p, method="BH")
limma_outcome_c18neg$p.fdr = p.adjust(limma_outcome_c18neg$p, method="BH")
limma_outcome_hilicpos$metabolite = colnames(hilicpos_MSMICA_featuretable_clean_t[, -c(1:6)])
limma_outcome_c18neg$metabolite = colnames(c18neg_MSMICA_featuretable_clean_t[, -c(1:6)])
# add Name, HMDB_ID, KEGG_ID, Mono_mass, Adduct, mz, time from hilicpos_MSMICA_identified_metabolites to limma_outcome_hilicpos
limma_outcome_hilicpos$Name = hilicpos_MSMICA_identified_metabolites$Name
limma_outcome_hilicpos$HMDB_ID = hilicpos_MSMICA_identified_metabolites$HMDB_ID
limma_outcome_hilicpos$KEGG_ID = hilicpos_MSMICA_identified_metabolites$KEGG_ID
limma_outcome_hilicpos$Mono_mass = hilicpos_MSMICA_identified_metabolites$Mono_mass
limma_outcome_hilicpos$Adduct = hilicpos_MSMICA_identified_metabolites$Adduct
limma_outcome_hilicpos$mz = hilicpos_MSMICA_identified_metabolites$mz
limma_outcome_hilicpos$time = hilicpos_MSMICA_identified_metabolites$time
limma_outcome_c18neg$Name = c18neg_MSMICA_identified_metabolites$Name
limma_outcome_c18neg$HMDB_ID = c18neg_MSMICA_identified_metabolites$HMDB_ID
limma_outcome_c18neg$KEGG_ID = c18neg_MSMICA_identified_metabolites$KEGG_ID
limma_outcome_c18neg$Mono_mass = c18neg_MSMICA_identified_metabolites$Mono_mass
limma_outcome_c18neg$Adduct = c18neg_MSMICA_identified_metabolites$Adduct
limma_outcome_c18neg$mz = c18neg_MSMICA_identified_metabolites$mz
limma_outcome_c18neg$time = c18neg_MSMICA_identified_metabolites$time
# Organize the column names: Name, HMDB_ID, KEGG_ID, Mono_mass, Adduct, mz, time, t_statistics, p, p.fdr, Cancer_log2FC
limma_outcome_hilicpos = limma_outcome_hilicpos %>%
select(Name, HMDB_ID, KEGG_ID, Mono_mass, Adduct, mz, time, t_statistic, p, p.fdr, Cancer_log2FC) %>%
arrange(p)
limma_outcome_c18neg = limma_outcome_c18neg %>%
select(Name, HMDB_ID, KEGG_ID, Mono_mass, Adduct, mz, time, t_statistic, p, p.fdr, Cancer_log2FC) %>%
arrange(p)
# select only the significant metabolites
limma_outcome_hilicpos_sig = limma_outcome_hilicpos %>%
filter(p < 0.05)
limma_outcome_c18neg_sig = limma_outcome_c18neg %>%
filter(p < 0.05)
# print the results
print(limma_outcome_hilicpos_sig, width=Inf)
## # A tibble: 476 × 11
## Name HMDB_ID KEGG_ID Mono_mass Adduct mz
## <chr> <chr> <chr> <dbl> <chr> <dbl>
## 1 N-Butylacetamide HMDB0031651 NA 115. M+H 116.
## 2 Iprovalicarb NA C18866 320. M+H 321.
## 3 N-Acetyl-1-methyl-L-histidine HMDB0240340 NA 211. M+H 212.
## 4 Lactose HMDB0000186 C00243 342. M+K 381.
## 5 3-Methylhistamine HMDB0001861 NA 125. M+H 126.
## 6 Avenestergenin A1 HMDB0035264 NA 637. M+H 638.
## 7 N-Acetyl-L-arginine HMDB0004620 NA 216. M+H 217.
## 8 Beta-hydroxyfentanyl NA C22749 352. M+H 353.
## 9 Cheritamine HMDB0034872 NA 593. M+H 594.
## 10 12-Hydroxyjasmonate sulfate NA C21671 306. M+H 307.
## time t_statistic p p.fdr Cancer_log2FC
## <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 42 -5.81 0.000000966 0.00117 -1.76
## 2 33 -7.04 0.00000118 0.00117 -3.12
## 3 81 -5.90 0.00000120 0.00117 -2.33
## 4 83 -5.61 0.00000184 0.00134 -1.24
## 5 89 -5.56 0.00000235 0.00137 -2.28
## 6 38 -5.35 0.00000488 0.00222 -2.99
## 7 67 -5.39 0.00000601 0.00222 -2.19
## 8 78 -5.23 0.00000611 0.00222 -2.46
## 9 39 -5.04 0.0000121 0.00341 -3.42
## 10 56 -5.00 0.0000134 0.00341 -2.87
## # ℹ 466 more rows
print(limma_outcome_c18neg_sig, width=Inf)
## # A tibble: 447 × 11
## Name HMDB_ID KEGG_ID
## <chr> <chr> <chr>
## 1 D-Glucuronic Acid HMDB0000127 C00191
## 2 Uridine HMDB0000296 C00299
## 3 Diphenol glucuronide HMDB0059998 NA
## 4 Guanosine HMDB0000133 C00387
## 5 Cytidine diphosphoramidate NA C22031
## 6 Erucic Acid HMDB0002068 C08316
## 7 N-(heptadecanoyl)-sphing-4-enine-1-phosphocholine HMDB0240609 NA
## 8 PS(22:0/22:4(7Z,10Z,13Z,16Z)) HMDB0112730 NA
## 9 Paullinic acid HMDB0035159 C21946
## 10 11-O-Demethylpradinone I NA C06775
## Mono_mass Adduct mz time t_statistic p p.fdr Cancer_log2FC
## <dbl> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 194. M-H 193. 21 -7.09 0.0000000207 0.0000503 -3.66
## 2 244. M-H 243. 29 -6.23 0.000000302 0.000255 -1.34
## 3 286. M+Cl 321. 24 -6.21 0.000000315 0.000255 -1.59
## 4 283. M-H 282. 27 -5.85 0.000000973 0.000546 -1.19
## 5 402. M-H 401. 22 -5.90 0.00000112 0.000546 -3.83
## 6 338. M-H 337. 286 5.54 0.00000257 0.000978 1.91
## 7 717. M-H 716. 263 5.51 0.00000282 0.000978 1.63
## 8 896. M-H 895. 285 5.97 0.00000348 0.00106 4.35
## 9 310. M-H 309. 263 5.29 0.00000559 0.00151 1.58
## 10 464. M-H 463. 20 -5.51 0.00000673 0.00164 -3.64
## # ℹ 437 more rows
# save the result as a csv file
write.csv(limma_outcome_hilicpos_sig, "MSMICA targeted metabolomics analysis/rcc_targeted_MSMICA_HILICpos_metabolites_limma_paired_t_test.csv", row.names = FALSE)
write.csv(limma_outcome_c18neg_sig, "MSMICA targeted metabolomics analysis/rcc_targeted_MSMICA_C18neg_metabolites_limma_paired_t_test.csv", row.names = FALSE)
MSMICA targeted PLS Discriminant Analysis (PLS-DA)
## Loading required package: MASS
##
## Attaching package: 'MASS'
## The following object is masked from 'package:dplyr':
##
## select
## Loading required package: lattice
## Warning: package 'lattice' was built under R version 4.3.3
## Loading required package: ggplot2
## Warning: package 'ggplot2' was built under R version 4.3.3
##
## Loaded mixOmics 6.26.0
## Thank you for using mixOmics!
## Tutorials: http://mixomics.org
## Bookdown vignette: https://mixomicsteam.github.io/Bookdown
## Questions, issues: Follow the prompts at http://mixomics.org/contact-us
## Cite us: citation('mixOmics')
set.seed(2024) # for reproducibility, remove for normal use
# This is the intensity data for the MSMICA identified metabolites in HILIC positive only
X = as.matrix(hilicpos_intensity)
# This is the sample type for each sample: normal or cancer
Y = factor(hilicpos_MSMICA_featuretable_clean_t$sample_type, levels = c("normal", "cancer"))
dim(X) # check the dimensions of the X dataframe
## [1] 42 2923
length(Y) # check the length of the Y dataframe
## [1] 42
# Preliminary Analysis with PCA
# run pca method on data (our intensity data was already log2 transformed, so we don't need to scale it here)
pca.RCC = pca(X, ncomp = 2, center = TRUE, scale = FALSE)
plot(pca.RCC) # barplot of the eigenvalues (explained variance per component)
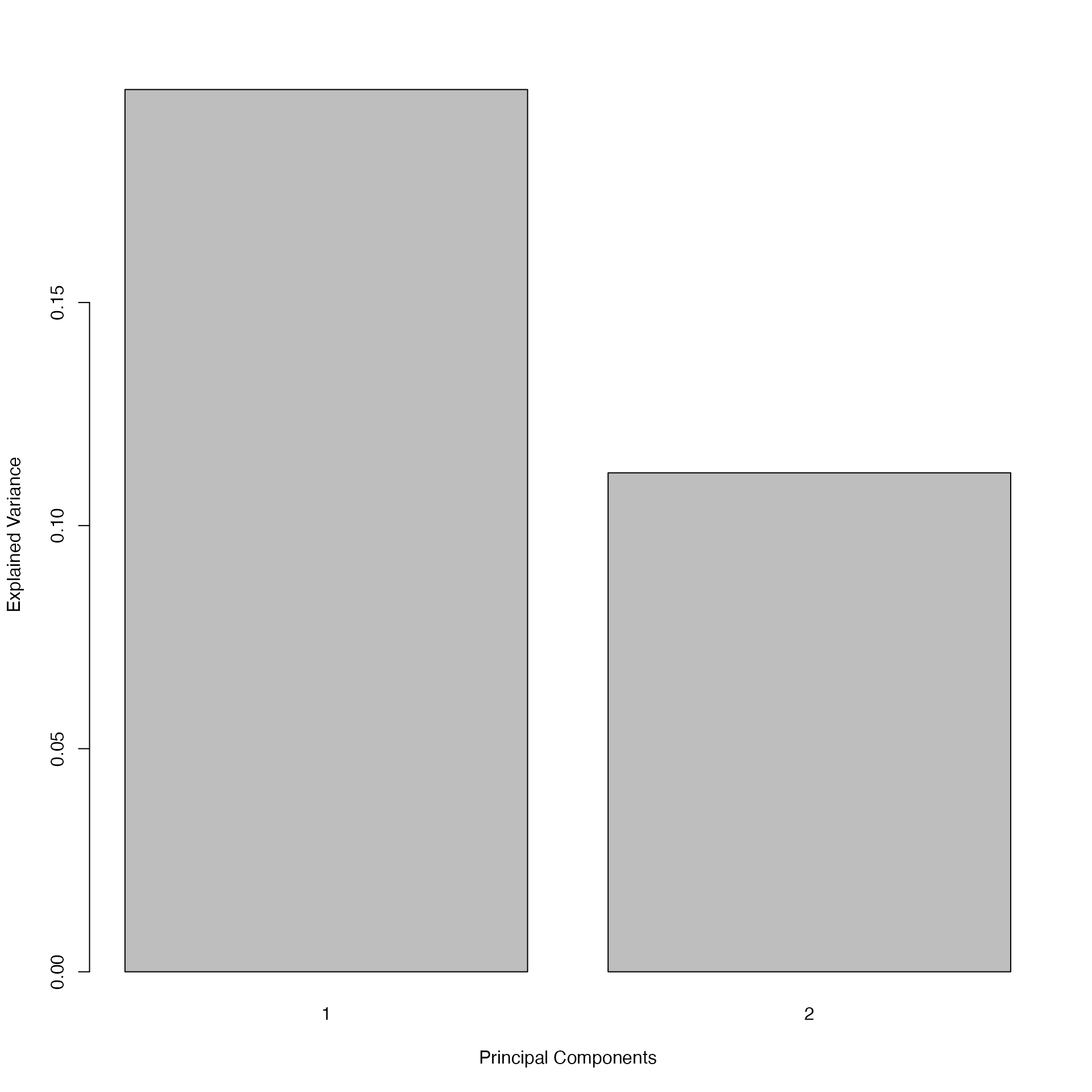
# Two components would be sufficient to explain a moderate proportion of the data's variance
plotIndiv(pca.RCC, group = Y, ind.names = TRUE, # plot the samples projected
legend = TRUE,
col.per.group = c("#F68B33", "#388ECC"), # set of colours
title = 'PCA on the RCC study, comp 1 & 2',
ellipse = TRUE) # onto the PCA
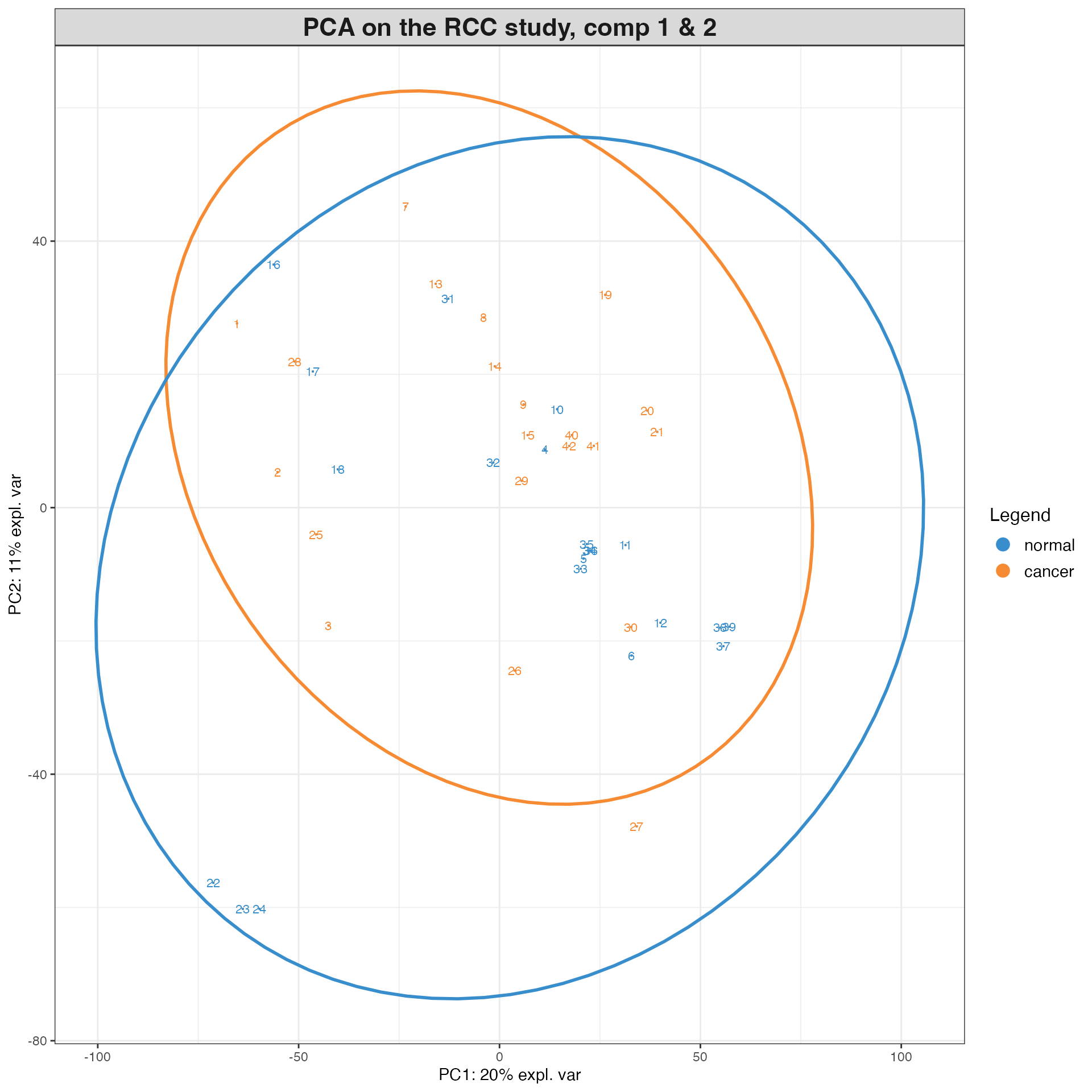
# It seems like only the normal tissue samples are separated from the cancer tissue samples. Cancer tissue samples are heterogenous. This is expected as the normal tissue samples are expected to have different metabolite profiles compared to the cancer tissue samples.
# PLS-DA - Note, PLS-DA is a supervised method, so we need to provide the class labels (Y) to the function. This is designed to find the metabolites that best discriminate between the two classes. Thus, it is not surprising that the PLS-DA model will perform better than the PCA model for class discrimination.
RCC_plsda <- plsda(X, Y, ncomp = 2) # set ncomp to 2 for performance assessment later
# plot the samples projected onto the first two components of the PLS-DA subspace
plotIndiv(RCC_plsda, comp = c(1,2), # plot samples from final model
group = Y, ind.names = TRUE, # colour by class label
ellipse = TRUE, legend = TRUE, # include 95% confidence ellipse,
col.per.group = c("#F68B33", "#388ECC"), # set of colours
title = 'Final PLS-DA on the RCC study, comp 1 & 2')
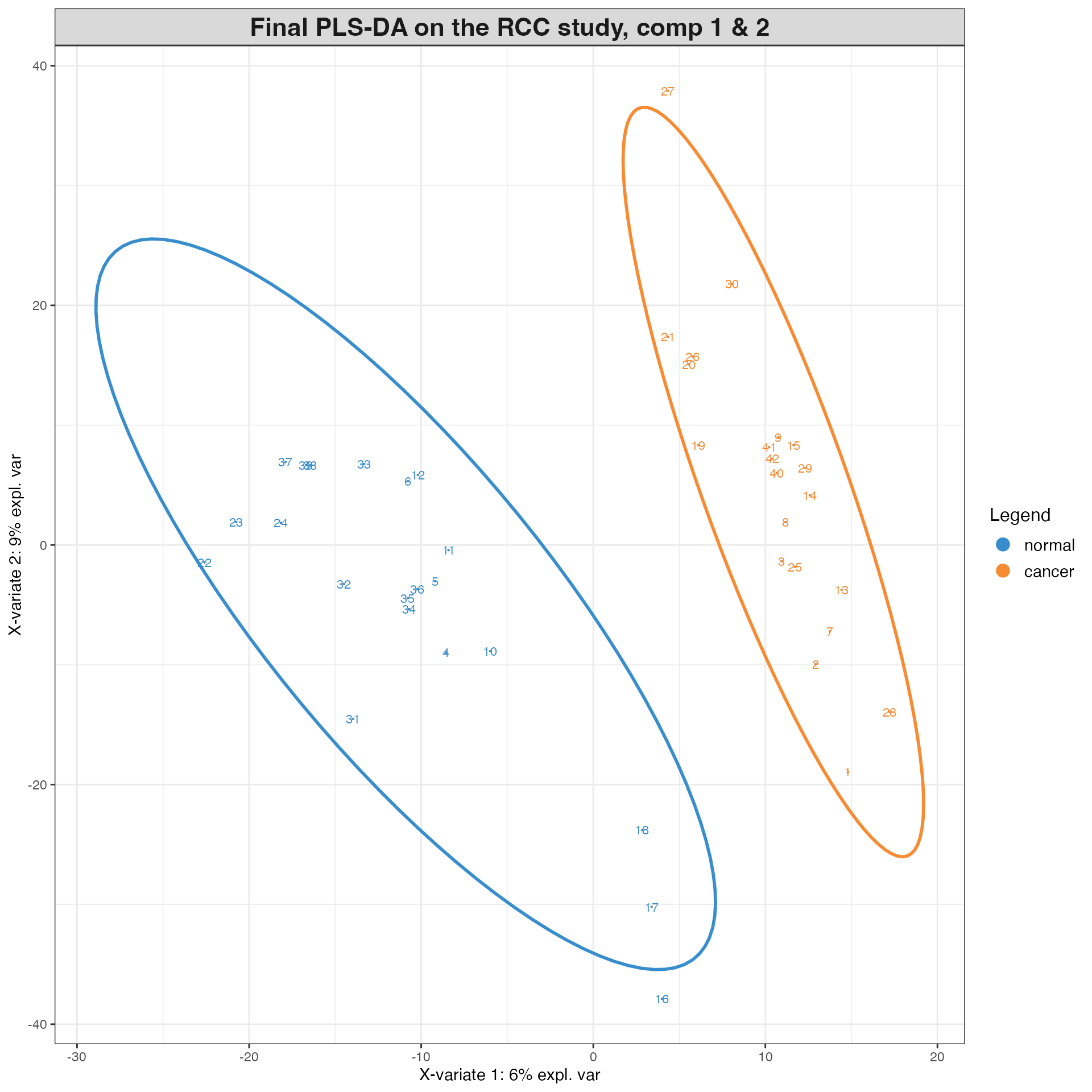
# list their VIP scores
# Obtain the VIP (Variable Importance in Projection) scores for the fitted PLS-DA model
VIP_scores <- mixOmics::vip(RCC_plsda)
# extract the VIP scores
VIP_scores = VIP_scores[, 1]
VIP_scores_df = tibble(
Name = names(VIP_scores),
VIP_scores = as.numeric(VIP_scores))
VIP_scores_df = VIP_scores_df %>%
arrange(desc(VIP_scores))
print(VIP_scores_df)
## # A tibble: 2,923 × 2
## Name VIP_scores
## <chr> <dbl>
## 1 1-Stearoyl-2-arachidonoyl-sn-glycero-3-phosphoserine 3.67
## 2 N-Butylacetamide 3.52
## 3 Guanosine 3.28
## 4 12-Hydroxyjasmonate sulfate 3.16
## 5 2-Hydroxy-1-(2,4,6-trihydroxyphenyl)-3-(3,4-dihydroxyphenyl)propa… 3.16
## 6 Avenestergenin A1 3.14
## 7 N-Acetyl-L-arginine 3.08
## 8 3-Methylhistamine 3.08
## 9 Lactose 3.02
## 10 Arctinone A acetate 3.01
## # ℹ 2,913 more rows
# save the VIP scores as a csv file
write.csv(VIP_scores_df, "MSMICA targeted metabolomics analysis/rcc_targeted_MSMICA_HILICpos_metabolites_PLS-DA_VIP_scores.csv", row.names = FALSE)
# Prepare the data for MSMICA untargeted metabolomics analysis ############################################################################################################
# HILIC positive ############################################################################################################
# all MSMICA identified metabolites in HILIC positive and C18 negative #############################
# transpose the table starting at column 13
feature_table_exp_hilicpos_imputed_t = t(feature_table_exp_hilicpos_imputed[,3:ncol(feature_table_exp_hilicpos_imputed)])
feature_table_exp_hilicpos_imputed_t = as.data.frame(feature_table_exp_hilicpos_imputed_t)
# set the column names as the combination of mz and time
colnames(feature_table_exp_hilicpos_imputed_t) = paste0(feature_table_exp_hilicpos_imputed$mz, "_", feature_table_exp_hilicpos_imputed$time)
# add Group_ID as a column
feature_table_exp_hilicpos_imputed_t$Group_ID = rownames(feature_table_exp_hilicpos_imputed_t)
# move Group_ID as the first column
feature_table_exp_hilicpos_imputed_t = tibble(feature_table_exp_hilicpos_imputed_t) %>%
dplyr::select(Group_ID, everything())
# Seperate the Group_ID into columns: Group, Temperature, Time, SampleID
feature_table_exp_hilicpos_imputed_t = feature_table_exp_hilicpos_imputed_t %>%
tidyr::separate(Group_ID, c("Group", "Temperature", "Time"), sep = "_") %>%
# create a new column, sample_tyoe, to indicate the sample type: if Group starts with NM, then it is normal, otherwise it is cancer
mutate(sample_type = ifelse(stringr::str_detect(Group, "^NM"), "normal", "cancer")) %>%
# move sample type to the first column
dplyr::select(sample_type, everything())
# separate the Group column into Group and ParticipantID: the first two characters are Group, the rest are ParticipantID
feature_table_exp_hilicpos_imputed_t = feature_table_exp_hilicpos_imputed_t %>%
tidyr::separate(Group, c("Group", "ParticipantID"), sep = 2)
# Convert all intensity values to log2
feature_table_exp_hilicpos_imputed_t[, -c(1:5)] = feature_table_exp_hilicpos_imputed_t[, -c(1:5)] %>%
mutate_all(~log2(.))
# check the data ready for MSMICA untargeted metabolomics analysis
print(feature_table_exp_hilicpos_imputed_t)
## # A tibble: 14 × 2,810
## sample_type Group ParticipantID Temperature Time `85.0284_49` `85.0477_83`
## <chr> <chr> <chr> <chr> <chr> <dbl> <dbl>
## 1 normal NM 11 GS 0 26.4 24.3
## 2 normal NM 4 GS 0 25.7 23.5
## 3 normal NM 5 GS 0 26.5 23.6
## 4 normal NM 6 GS 0 25.5 24.1
## 5 normal NM 7 GS 0 25.6 22.9
## 6 normal NM 8 GS 0 26.0 24.0
## 7 normal NM 9 GS 0 24.6 23.3
## 8 cancer TM 11 GS 0 26.6 23.6
## 9 cancer TM 4 GS 0 24.8 23.3
## 10 cancer TM 5 GS 0 26.6 23.0
## 11 cancer TM 6 GS 0 26.6 24.6
## 12 cancer TM 7 GS 0 26.4 24.8
## 13 cancer TM 8 GS 0 25.7 21.3
## 14 cancer TM 9 GS 0 25.3 23.8
## # ℹ 2,803 more variables: `85.0841_109` <dbl>, `86.06_86` <dbl>,
## # `86.0964_48` <dbl>, `86.9086_63` <dbl>, `86.9504_32` <dbl>,
## # `87.0441_293` <dbl>, `87.0553_86` <dbl>, `87.0619_32` <dbl>,
## # `87.0917_284` <dbl>, `87.0998_48` <dbl>, `87.174_33` <dbl>,
## # `87.2858_33` <dbl>, `87.3982_35` <dbl>, `87.5098_35` <dbl>,
## # `88.0216_281` <dbl>, `88.0393_279` <dbl>, `88.0393_83` <dbl>,
## # `88.0578_79` <dbl>, `88.0757_86` <dbl>, `88.9526_258` <dbl>, …
# C18 negative ############################################################################################################
# all MSMICA identified metabolites #############################
feature_table_exp_c18neg_imputed_t = t(feature_table_exp_c18neg_imputed[,3:ncol(feature_table_exp_c18neg_imputed)])
feature_table_exp_c18neg_imputed_t = as.data.frame(feature_table_exp_c18neg_imputed_t)
# set the column names as the combination of mz and time
colnames(feature_table_exp_c18neg_imputed_t) = paste0(feature_table_exp_c18neg_imputed$mz, "_", feature_table_exp_c18neg_imputed$time)
# add Group_ID as a column
feature_table_exp_c18neg_imputed_t$Group_ID = rownames(feature_table_exp_c18neg_imputed_t)
# move Group_ID as the first column
feature_table_exp_c18neg_imputed_t = tibble(feature_table_exp_c18neg_imputed_t) %>%
dplyr::select(Group_ID, everything())
# Seperate the Group_ID into columns: Group, Temperature, Time, SampleID
feature_table_exp_c18neg_imputed_t = feature_table_exp_c18neg_imputed_t %>%
separate(Group_ID, c("Group", "Temperature", "Time"), sep = "_") %>%
# create a new column, sample_tyoe, to indicate the sample type: if Group starts with NM, then it is normal, otherwise it is cancer
mutate(sample_type = ifelse(stringr::str_detect(Group, "^NM"), "normal", "cancer")) %>%
# move sample type to the first column
dplyr::select(sample_type, everything())
# separate the Group column into Group and ParticipantID: the first two characters are Group, the rest are ParticipantID
feature_table_exp_c18neg_imputed_t = feature_table_exp_c18neg_imputed_t %>%
separate(Group, c("Group", "ParticipantID"), sep = 2)
# Convert all intensity values to log2
feature_table_exp_c18neg_imputed_t[, -c(1:5)] = feature_table_exp_c18neg_imputed_t[, -c(1:5)] %>%
mutate_all(~log2(.))
# check the data ready for MSMICA untargeted metabolomics analysis
print(feature_table_exp_c18neg_imputed_t)
## # A tibble: 14 × 4,220
## sample_type Group ParticipantID Temperature Time `85.0044_123` `85.0044_288`
## <chr> <chr> <chr> <chr> <chr> <dbl> <dbl>
## 1 normal NM 11 GS 0 22.9 22.1
## 2 normal NM 4 GS 0 23.3 21.9
## 3 normal NM 5 GS 0 22.0 21.6
## 4 normal NM 6 GS 0 22.4 21.6
## 5 normal NM 7 GS 0 23.0 21.9
## 6 normal NM 8 GS 0 21.1 21.7
## 7 normal NM 9 GS 0 20.9 21.6
## 8 cancer TM 11 GS 0 23.7 22.0
## 9 cancer TM 4 GS 0 19.8 21.8
## 10 cancer TM 5 GS 0 23.5 22.0
## 11 cancer TM 6 GS 0 23.5 22.1
## 12 cancer TM 7 GS 0 23.4 21.8
## 13 cancer TM 8 GS 0 23.4 21.2
## 14 cancer TM 9 GS 0 22.6 21.7
## # ℹ 4,213 more variables: `85.0295_293` <dbl>, `85.0295_25` <dbl>,
## # `85.0295_95` <dbl>, `85.3585_236` <dbl>, `85.7258_217` <dbl>,
## # `86.0248_292` <dbl>, `86.0248_91` <dbl>, `86.1808_294` <dbl>,
## # `87.0087_24` <dbl>, `87.02_289` <dbl>, `87.0273_34` <dbl>,
## # `87.0452_30` <dbl>, `87.4819_227` <dbl>, `87.5205_173` <dbl>,
## # `87.8345_243` <dbl>, `88.004_198` <dbl>, `88.0404_29` <dbl>,
## # `88.988_32` <dbl>, `89.0088_23` <dbl>, `89.0194_24` <dbl>, …
############################# limma paired t-test #############################
# HILIC positive ############################################################################################################
# Factorize the samples into normal and cancer
Group_hilicpos = factor(feature_table_exp_hilicpos_imputed_t$sample_type, levels = c("normal", "cancer"))
# Factorize the ParticipantID
ParticipantID_hilicpos = factor(feature_table_exp_hilicpos_imputed_t$ParticipantID)
# Create the design matrix
design_T_hilicpos <- model.matrix(~ ParticipantID_hilicpos+Group_hilicpos)
# Extract the intensity values for the limma analysis
hilicpos_intensity = feature_table_exp_hilicpos_imputed_t[, 6:ncol(feature_table_exp_hilicpos_imputed_t)]
hilicpos_intensity = as.data.frame(hilicpos_intensity)
# transpose the metabolite intensity data
y_T_hilicpos = t(hilicpos_intensity)
# Fit the linear model
fit_hilicpos = lmFit(y_T_hilicpos, design_T_hilicpos)
fit2_hilicpos <- eBayes(fit_hilicpos)
# chcek the top table in the limma paited t-test output
topTable(fit2_hilicpos, coef="Group_hilicposcancer", adjust="BH")
## logFC AveExpr t P.Value adj.P.Val B
## 381.0789_83 -1.294320 25.34860 -4.126351 0.002771802 0.9989403 -4.484285
## 387.0549_178 -1.559068 21.73473 -3.999655 0.003337587 0.9989403 -4.487121
## 242.1749_28 -2.066738 22.05930 -3.806452 0.004449652 0.9989403 -4.491727
## 386.238_27 -2.618954 22.10443 -3.758988 0.004779116 0.9989403 -4.492913
## 559.5185_31 -3.849687 21.31891 -3.624924 0.005856884 0.9989403 -4.496382
## 264.1957_34 -2.832936 22.93908 -3.601827 0.006067149 0.9989403 -4.496998
## 117.1103_42 -3.106604 22.83724 -3.535649 0.006714842 0.9989403 -4.498793
## 465.0513_214 1.757063 22.08679 3.495121 0.007147086 0.9989403 -4.499915
## 442.3889_29 2.453958 22.07238 3.460156 0.007543503 0.9989403 -4.500897
## 599.5033_29 -2.350697 21.55573 -3.441356 0.007766128 0.9989403 -4.501430
# Extract all relevant values from the limma output
limma_outcome_hilicpos = data.frame(
Cancer_log2FC = c(fit2_hilicpos$coefficients[, "Group_hilicposcancer"]),
t_statistic = c(fit2_hilicpos$t[, "Group_hilicposcancer"]),
p = c(fit2_hilicpos$p.value[, "Group_hilicposcancer"]))
limma_outcome_hilicpos$p.fdr = p.adjust(limma_outcome_hilicpos$p, method="BH")
limma_outcome_hilicpos$mz_time = colnames(feature_table_exp_hilicpos_imputed_t[, -c(1:5)])
limma_outcome_hilicpos = tibble(limma_outcome_hilicpos)
# mz, time from feature_table_exp_hilicpos_imputed to limma_outcome_hilicpos
limma_outcome_hilicpos$mz = feature_table_exp_hilicpos_imputed$mz
limma_outcome_hilicpos$time = feature_table_exp_hilicpos_imputed$time
# Organize the column names: mz, time, t_statistics, p, p.fdr, Cancer_log2FC
limma_outcome_hilicpos = limma_outcome_hilicpos %>%
dplyr::select(mz, time, t_statistic, p, p.fdr, Cancer_log2FC) %>%
arrange(p) %>%
tibble()
# select only the significant metabolites
limma_outcome_hilicpos_sig_untargeted = limma_outcome_hilicpos %>%
filter(p < 0.05)
# left join the MSMICA results to the limma_outcome_hilicpos_sig based on mz and time
limma_outcome_hilicpos_sig_untargeted_MSMICA = left_join(limma_outcome_hilicpos_sig_untargeted, hilicpos_MSMICA_identified_metabolites, by=c("mz"="mz", "time"="time"))
# print the results
print(limma_outcome_hilicpos_sig_untargeted_MSMICA)
## # A tibble: 87 × 77
## mz time t_statistic p p.fdr Cancer_log2FC mz_time Adduct Name
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr>
## 1 381. 83 -4.13 0.00277 0.999 -1.29 381.0789_83 M+K Lacto…
## 2 387. 178 -4.00 0.00334 0.999 -1.56 NA NA NA
## 3 242. 28 -3.81 0.00445 0.999 -2.07 242.1749_28 M+H Valer…
## 4 386. 27 -3.76 0.00478 0.999 -2.62 NA NA NA
## 5 560. 31 -3.62 0.00586 0.999 -3.85 NA NA NA
## 6 264. 34 -3.60 0.00607 0.999 -2.83 264.1957_34 M+H Desve…
## 7 117. 42 -3.54 0.00671 0.999 -3.11 NA NA NA
## 8 465. 214 3.50 0.00715 0.999 1.76 NA NA NA
## 9 442. 29 3.46 0.00754 0.999 2.45 442.3889_29 M+H Nonad…
## 10 600. 29 -3.44 0.00777 0.999 -2.35 NA NA NA
## # ℹ 77 more rows
## # ℹ 68 more variables: identification_type <chr>, identification_method <chr>,
## # Probability <dbl>, metabolite_within_feature_number <dbl>,
## # isomer_exist <lgl>, time_predicted <dbl>, time_difference <dbl>,
## # mean_intensity <dbl>, PubChem_compound_id <dbl>, HMDB_ID <chr>,
## # KEGG_ID <chr>, LIPID_MAPS_ID <chr>, ChEBI_ID <dbl>, BioCyc_ID <chr>,
## # DrugBank_ID <chr>, Mono_mass <dbl>, Formula <chr>, Charge <dbl>, …
# save the result as a csv file
write.csv(limma_outcome_hilicpos_sig_untargeted_MSMICA, "MSMICA untargeted metabolomics analysis/rcc_MSMICA_untargeted_hilicpos_metabolites_limma_paired_t_test.csv", row.names = FALSE)
# C18 negative ############################################################################################################
# Factorize the samples into normal and cancer
Group_c18neg = factor(feature_table_exp_c18neg_imputed_t$sample_type, levels = c("normal", "cancer"))
# Factorize the ParticipantID
ParticipantID_c18neg = factor(feature_table_exp_c18neg_imputed_t$ParticipantID)
# Create the design matrix
design_T_c18neg <- model.matrix(~ ParticipantID_c18neg+Group_c18neg)
# Extract the intensity values for the limma analysis
c18neg_intensity = feature_table_exp_c18neg_imputed_t[, 6:ncol(feature_table_exp_c18neg_imputed_t)]
# transpose the metabolite intensity data
y_T_c18neg = t(c18neg_intensity)
# Fit the linear model
fit_c18neg = lmFit(y_T_c18neg, design_T_c18neg)
fit2_c18neg <- eBayes(fit_c18neg)
# chcek the top table in the limma paited t-test output
topTable(fit2_c18neg, coef="Group_c18negcancer", adjust="BH")
## logFC AveExpr t P.Value adj.P.Val B
## 154.9656_21 -2.882124 22.38869 -6.744578 0.0001101287 0.4641923 -4.189543
## 716.5808_263 1.764388 20.59766 4.949691 0.0009376188 0.8898829 -4.247140
## 466.8895_80 2.005374 19.77082 4.868171 0.0010442325 0.8898829 -4.250799
## 868.6075_282 1.696961 20.85049 4.799984 0.0011435091 0.8898829 -4.253949
## 664.8299_69 -1.195280 21.21145 -4.739879 0.0012395136 0.8898829 -4.256797
## 209.0815_31 1.539345 21.68307 4.389639 0.0020037052 0.8898829 -4.274795
## 221.0123_29 1.417734 23.97016 4.069227 0.0031584377 0.8898829 -4.293575
## 403.2516_246 1.681656 22.53564 3.949546 0.0037578698 0.8898829 -4.301216
## 487.0009_61 -1.732563 21.04422 -3.888754 0.0041078412 0.8898829 -4.305236
## 310.283_263 1.594794 23.80621 3.872067 0.0042098608 0.8898829 -4.306356
# Extract all relevant values from the limma output
limma_outcome_c18neg = data.frame(
Cancer_log2FC = c(fit2_c18neg$coefficients[, "Group_c18negcancer"]),
t_statistic = c(fit2_c18neg$t[, "Group_c18negcancer"]),
p = c(fit2_c18neg$p.value[, "Group_c18negcancer"]))
limma_outcome_c18neg$p.fdr = p.adjust(limma_outcome_c18neg$p, method="BH")
limma_outcome_c18neg$mz_time = colnames(feature_table_exp_c18neg_imputed_t[, -c(1:5)])
limma_outcome_c18neg = tibble(limma_outcome_c18neg)
# mz, time from feature_table_exp_c18neg_imputed to limma_outcome_c18neg
limma_outcome_c18neg$mz = feature_table_exp_c18neg_imputed$mz
limma_outcome_c18neg$time = feature_table_exp_c18neg_imputed$time
# Organize the column names: mz, time, t_statistics, p, p.fdr, Cancer_log2FC
limma_outcome_c18neg = limma_outcome_c18neg %>%
dplyr::select(mz, time, t_statistic, p, p.fdr, Cancer_log2FC) %>%
arrange(p) %>%
tibble()
# select only the significant metabolites
limma_outcome_c18neg_sig_untargeted = limma_outcome_c18neg %>%
filter(p < 0.05)
# left join the MSMICA results to the limma_outcome_c18neg_sig based on mz and time
limma_outcome_c18neg_sig_untargeted_MSMICA = left_join(limma_outcome_c18neg_sig_untargeted, c18neg_MSMICA_identified_metabolites, by=c("mz"="mz", "time"="time"))
# print the results
print(limma_outcome_c18neg_sig_untargeted_MSMICA)
## # A tibble: 208 × 77
## mz time t_statistic p p.fdr Cancer_log2FC mz_time Adduct Name
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr>
## 1 155. 21 -6.74 0.000110 0.464 -2.88 NA NA NA
## 2 717. 263 4.95 0.000938 0.890 1.76 NA NA NA
## 3 467. 80 4.87 0.00104 0.890 2.01 NA NA NA
## 4 869. 282 4.80 0.00114 0.890 1.70 868.6075_2… M-H PS(2…
## 5 665. 69 -4.74 0.00124 0.890 -1.20 NA NA NA
## 6 209. 31 4.39 0.00200 0.890 1.54 209.0815_31 M-H 3,5-…
## 7 209. 31 4.39 0.00200 0.890 1.54 209.0815_31 M-H Phos…
## 8 221. 29 4.07 0.00316 0.890 1.42 NA NA NA
## 9 403. 246 3.95 0.00376 0.890 1.68 403.2516_2… M-H 7alp…
## 10 487. 61 -3.89 0.00411 0.890 -1.73 NA NA NA
## # ℹ 198 more rows
## # ℹ 68 more variables: identification_type <chr>, identification_method <chr>,
## # Probability <dbl>, metabolite_within_feature_number <dbl>,
## # isomer_exist <lgl>, time_predicted <dbl>, time_difference <dbl>,
## # mean_intensity <dbl>, PubChem_compound_id <dbl>, HMDB_ID <chr>,
## # KEGG_ID <chr>, LIPID_MAPS_ID <chr>, ChEBI_ID <dbl>, BioCyc_ID <chr>,
## # DrugBank_ID <chr>, Mono_mass <dbl>, Formula <chr>, Charge <dbl>, …
# save the result as a csv file
write.csv(limma_outcome_c18neg_sig_untargeted_MSMICA, "MSMICA untargeted metabolomics analysis/rcc_MSMICA_untargeted_c18neg_metabolites_limma_paired_t_test.csv", row.names = FALSE)